The TheraRadar Brief
Every drug has multiple stories. Most never get told.
TL1A: The Target Behind the $18 Billion Bet
Every approved IBD drug fights inflammation. None of them touch fibrosis. TL1A does both.
Get the next TheraRadar Brief in your inbox
Drug development, biology, and market dynamics — free, every week.
30% of Crohn's disease patients will need surgery within ten years of diagnosis. Not because their drugs failed to control inflammation - but because no approved drug stops the scar tissue that narrows their bowel until it blocks.
TL1A is the first target that could change that.
What is TL1A?
TL1A (TNF-like ligand 1A) is a cytokine - a signaling protein - encoded by the TNFSF15 gene. It belongs to the TNF superfamily, the same family as TNF-alpha, which is the target of Humira, Remicade, and every other anti-TNF drug on the market.
But TL1A does something TNF-alpha does not. It drives both inflammation and fibrosis - the structural scarring that narrows the intestinal wall and eventually requires surgery. TNF blockers quiet the inflammation. TL1A blockers could address the damage itself.
Three players, one system
The TL1A signaling system has three components:
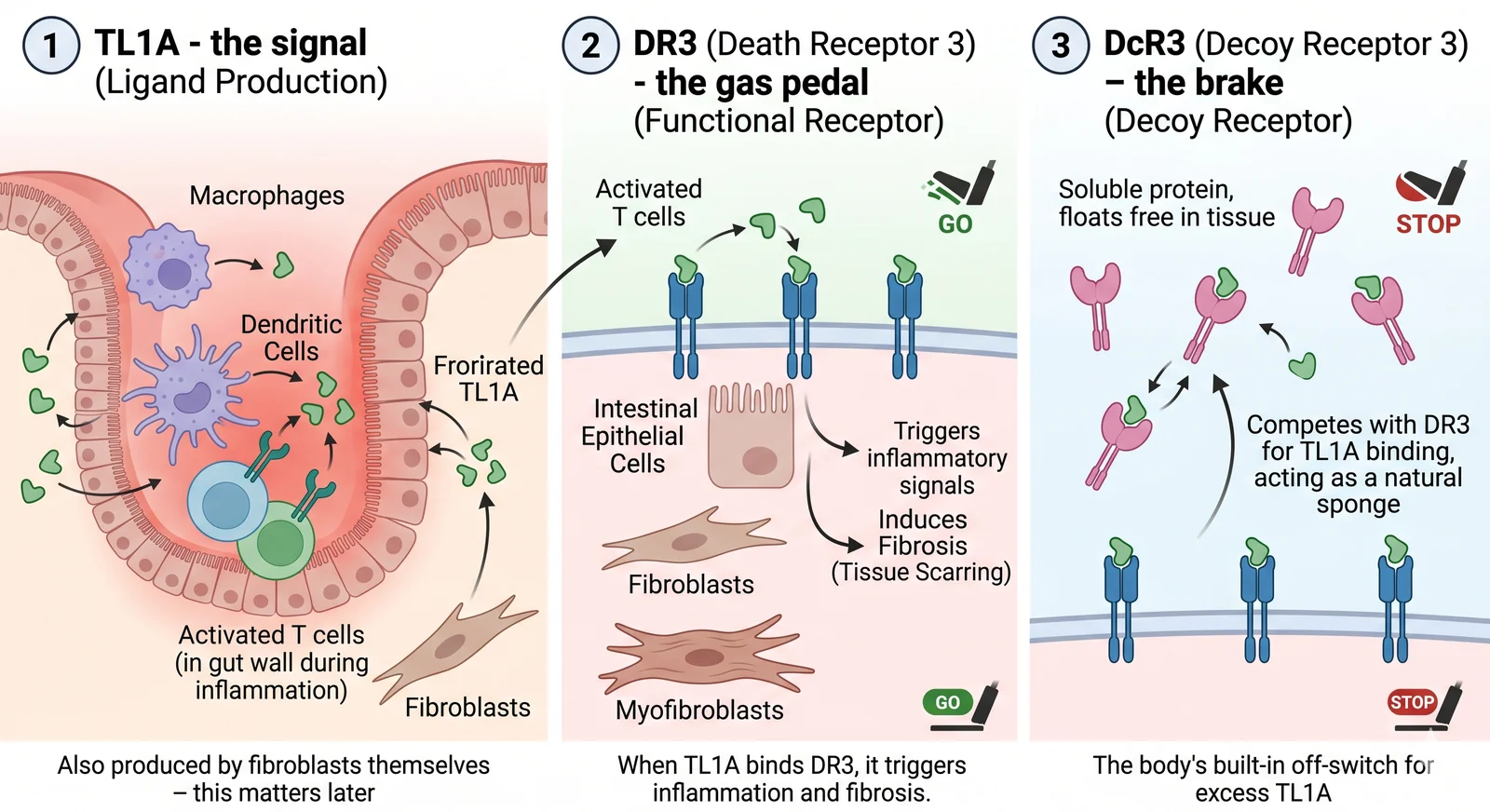
TheraRadar / NanoBanana. Sources: Dalal & Bhatt, PMC (2025); Shih & Bhatt, Scientific Reports (2020)
TL1A - the signal
Produced by macrophages, dendritic cells, and activated T cells in the gut wall during inflammation. Also produced by fibroblasts themselves - this matters later.
DR3 (Death Receptor 3) - the gas pedal
The functional receptor. Found on activated T cells, intestinal epithelial cells, and - critically - on fibroblasts and myofibroblasts. When TL1A binds DR3, it triggers inflammation and fibrosis.
DcR3 (Decoy Receptor 3) - the brake
A soluble protein (no transmembrane domain - it floats free in tissue). Competes with DR3 for TL1A binding, acting as a natural sponge. The body's built-in off-switch for excess TL1A.
What happens when TL1A binds DR3
When TL1A engages the DR3 receptor, two things happen simultaneously - and this is what makes the target unique.
The inflammatory arm. On T cells, TL1A-DR3 signaling recruits adaptor proteins (TRAF, RIPK1) that activate the NF-kB transcription factor - a master switch for inflammation. This triggers production of IL-1B, IL-23, IFN-gamma, and IL-17A. It amplifies existing immune activation rather than starting it from scratch. TL1A is a volume knob, not an on/off switch.
The fibrotic arm. On fibroblasts, TL1A-DR3 signaling activates a different set of pathways - Rho signaling and the TGF-B1/Smad3 cascade. These drive fibroblast-to-myofibroblast differentiation: the cells that lay down collagen and build scar tissue. The extracellular matrix thickens, the bowel wall stiffens, and over years, the lumen narrows into a stricture.
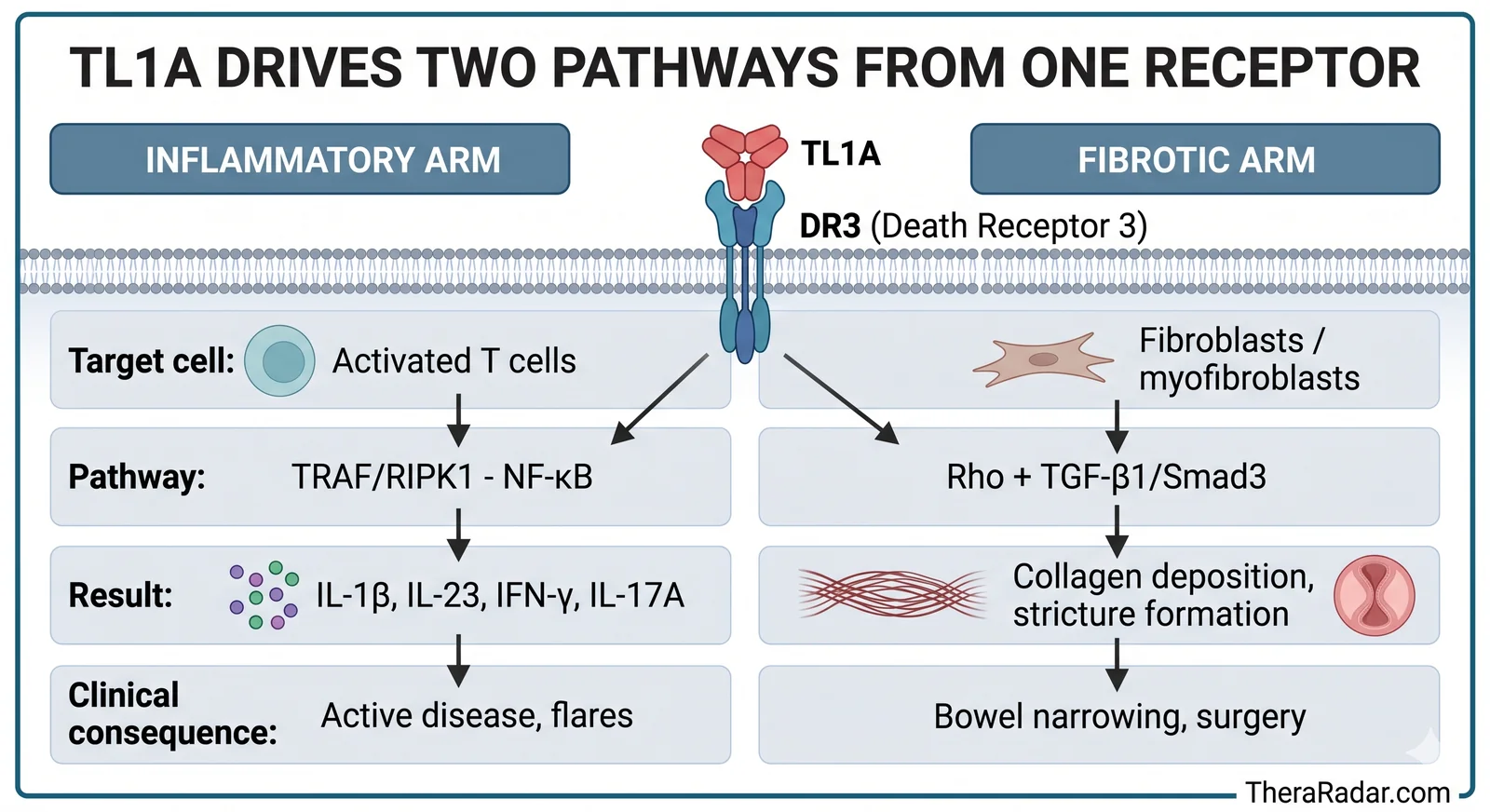
TheraRadar / NanoBanana
| Inflammatory arm | Fibrotic arm | |
|---|---|---|
| Target cell | Activated T cells | Fibroblasts / myofibroblasts |
| Pathway | TRAF/RIPK1 - NF-kB | Rho + TGF-B1/Smad3 |
| Result | IL-1B, IL-23, IFN-gamma, IL-17A | Collagen deposition, stricture formation |
| Clinical consequence | Active disease, flares | Bowel narrowing, surgery |
The loop that no current drug breaks
Here is the critical insight that explains why fibrosis progresses even in patients whose inflammation is well controlled.
When inflammatory signals activate macrophages in the gut wall, those macrophages produce TL1A. TL1A activates T cells (more inflammation) and fibroblasts (fibrosis begins). But activated fibroblasts don't just lay down scar tissue - they produce more TL1A themselves.
This creates an autocrine amplification loop. The fibroblasts signal to each other, independent of the original inflammatory trigger. Even after anti-TNF or anti-IL-23 therapy quiets the immune response, the fibroblast-to-fibroblast TL1A loop can keep running. Scar tissue keeps building. The bowel keeps narrowing.
This is why patients on effective anti-inflammatory biologics can still progress to surgery. The drugs they're taking were never designed to reach the fibrotic arm of the pathway. The TL1A blockade effect is independent of, and upstream and divergent from, TNF neutralization.
Want the next brief? Subscribe - free, every week.
Why existing IBD drugs can't touch fibrosis
| Drug class | Example | Mechanism | Inflammation | Fibrosis |
|---|---|---|---|---|
| TNF blocker | Humira, Remicade | Neutralize TNF-alpha | Yes | No |
| IL-23 blocker | Skyrizi, Tremfya | Block IL-23 cytokine | Yes | No |
| Integrin blocker | Entyvio | Block immune cell trafficking | Yes | No |
| JAK inhibitor | Rinvoq, Xeljanz | Suppress JAK signaling broadly | Yes | No |
| S1P modulator | Zeposia | Trap lymphocytes in lymph nodes | Yes | No |
| TL1A blocker | Phase 3 | Block TL1A-DR3 signaling | Yes | Yes |
TL1A blockers are the first IBD drug class designed to address both pathological arms of the disease.
The genetic signal
The case for TL1A isn't just mechanistic - it's genetic. Large-scale genome-wide association studies have identified over 200 IBD susceptibility loci, and TNFSF15 is consistently among them. A trans-ancestry GWAS meta-analysis of 86,640 European and East Asian subjects confirmed TNFSF15 as an IBD risk locus across populations, with particularly large effect sizes in East Asian patients.
Functional studies have shown that IBD-associated TNFSF15 haplotypes alter TL1A expression in monocytes and macrophages, linking genetic risk directly to the biological pathway. Patients carrying high-expression variants have more TL1A signaling, more inflammation, and - crucially - more fibrotic disease.
This genetic link is what made Prometheus Biosciences worth $10.8 billion to Merck. Prometheus didn't just have a TL1A-blocking antibody (tulisokibart). They had built a companion diagnostic around TNFSF15 genetics - a test that could identify which patients have TL1A-driven disease and are most likely to respond. In their Phase 2 trial (ARTEMIS-UC), biomarker-positive patients showed meaningfully better outcomes than the overall population. That's the precision medicine play: don't just block TL1A in everyone - find the patients where TL1A is actually driving the disease. Possibly one of the firsts in immunology drug discovery to use genetic biomarkers for patient selection. It will be really interesting to dig into what SNPs and polygenic risk score model they used.
The preclinical foundation
In preclinical studies, mice engineered to overexpress TL1A spontaneously developed intestinal strictures - the same fibrostenotic complications that drive surgery in Crohn's patients. The bowel wall thickened with collagen, the muscle layer hypertrophied, and the lumen narrowed.
Then came the result that built the scientific rationale. When researchers administered a neutralizing anti-TL1A antibody to mice with established colonic fibrosis, the fibrosis reversed - back to pre-inflammatory levels. Not just halted. Reversed. Collagen deposition dropped. Connective tissue growth factor and other fibrogenic mediators were suppressed. No other IBD target had demonstrated fibrosis reversal in vivo.
From preclinical fibrosis reversal to positive Phase 2 results in patients took roughly a decade. The $18 billion in deals followed.
The bigger picture: sequential immunotherapy
TL1A fits into a broader idea taking shape in autoimmune drug discovery. In a 2024 Nature Reviews Drug Discovery review, Ramírez-Valle et al. proposed sequential immunotherapy - a three-step framework: suppress inflammation, reset immune memory, then restore homeostasis and repair tissue. The paper explicitly names anti-TL1A as a future strategy. Early clinical evidence for the broader framework comes from CD19 CAR-T therapy in lupus, where patients achieved drug-free remission - but this remains early-stage, and nothing comparable has been tested in IBD yet.
What makes TL1A interesting in this context: most IBD drugs live entirely in Step 1 (suppress inflammation). TL1A bridges Steps 1 and 3 - it controls inflammation through NF-kB, but its anti-fibrotic action is closer to the tissue repair that Step 3 demands. Whether this framework proves out in IBD is an open question. But TL1A is the kind of target it was designed to find.
What we don't know yet
Anti-fibrotic drugs exist in other organs - pirfenidone and nintedanib are FDA-approved for lung fibrosis (IPF). But no anti-fibrotic therapy has ever been proven to work in the gut. The IBD Phase 2 trials measured standard inflammatory endpoints - clinical remission, endoscopic improvement - not fibrosis-specific outcomes. The Phase 3 programs will need to show not just that TL1A blockers reduce inflammation (they clearly do), but that they meaningfully impact the intestinal fibrosis that current drugs miss.
Three companies - Merck, Roche, and Sanofi/Teva - are running Phase 3 programs with readouts expected between 2026 and 2028. If the fibrosis signal holds in humans, TL1A blockers won't just be another option in IBD. They'll be a different category of drug. If it doesn't, $18 billion was spent on the most expensive mechanistic hypothesis in gastroenterology history.
That's the biology. In an upcoming brief, we'll follow the money - how three pharma giants committed $18 billion on this target within a single year, why Pfizer had one of these drugs for eight years and let it go, and what the Phase 3 readouts mean for the competitive landscape.
Go deeper with Pro
Pro gives you the data — patent cliffs, trial analytics, competitive landscapes, revenue tracking. Briefs tell the story with the data. Launch pricing: $99/month.
See Pro plansGet the next Brief in your inbox — free
Every drug has multiple stories. Most never get told. One brief every week. No spam.
Archive at /briefs/. Unsubscribe anytime.
Sources
- TL1A expression in IBD: Bamias et al., Expression, Localization, and Functional Activity of TL1A - Journal of Immunology (2003)
- DR3 in T cells: The ever-expanding role of cytokine receptor DR3 in T cells - PMC (2024)
- DcR3 structure: Decoy Strategies: The Structure of TL1A:DcR3 Complex - PMC (2011)
- Fibrosis mechanism: Shih et al., Direct signaling of TL1A-DR3 on fibroblasts induces intestinal fibrosis in vivo - Scientific Reports (2020)
- TGF-B1/Smad3 pathway: TL1A Promotes Fibrogenesis in Colonic Fibroblasts - Current Medical Science (2024)
- TL1A independent of TNF: Anti-TL1A Therapy Targets Tissue Inflammation and Fibrosis Pathways in UC - Inflammatory Bowel Diseases (2022)
- Mucosal immunity review: TL1A and DR3: A Co-stimulatory System in Gut Mucosal Immunity - Frontiers in Immunology (2019)
- TNFSF15 GWAS: Liu et al., Association analyses identify 38 susceptibility loci for IBD across populations - Nature Genetics (2015)
- TNFSF15 functional genetics: Siakavellas et al., Reduced monocyte TNFSF15/TL1A expression in IBD susceptibility - PLOS Genetics (2018)
- TL1A overexpression model: Inflammation independent TL1A-mediated Intestinal Fibrosis - Mucosal Immunology (2018)
- Fibrosis reversal: Inhibition of TL1A reverses established colonic fibrosis - Mucosal Immunology (2014)
- TL1A/DR3 review: Targeting TL1A and DR3: the new frontier of anti-cytokine therapy in IBD - PMC (2025)
- Sequential immunotherapy: Ramírez-Valle et al., Sequential immunotherapy: towards cures for autoimmunity - Nature Reviews Drug Discovery (2024)
- Merck acquisition: Merck Completes Acquisition of Prometheus Biosciences, Inc. (2023)
Spot an error? Reach out at hello@theraradar.com.