The TheraRadar Brief
Every drug has multiple stories. Most never get told.
Thalidomide Accidentally Invented Targeted Protein Degradation
Nobody knew why it caused birth defects for 50 years. The answer launched a new era of drug design.
Get the next TheraRadar Brief in your inbox
Drug development, biology, and market dynamics — free, every week.
In 1961, thalidomide was pulled from markets worldwide after causing severe birth defects in over 10,000 babies. It became the most infamous molecule in pharmaceutical history.
For the next fifty years, nobody could explain how it did it.
Get briefs like this every week - subscribe free.
The disaster
On October 1, 1957, the German pharmaceutical company Chemie Grunenthal launched a new sedative called Contergan. It was marketed as a safe sleeping pill - so safe it was available over the counter, and widely prescribed to pregnant women for morning sickness. Within four years it was sold in 46 countries under dozens of brand names.
By 1961, pediatricians in Germany and Australia independently noticed a surge in babies born with phocomelia - severely shortened or absent limbs. German pediatrician Widukind Lenz and Australian obstetrician William McBride traced the defects to thalidomide. The drug was withdrawn on November 26, 1961. Over 10,000 babies in 46 countries were born with severe malformations. Many more died before birth.
What made it devastating was the dose. According to Vargesson (2015), a single 50 mg tablet taken during a 16-day window - days 20 to 36 after fertilization - was sufficient to cause birth defects in up to 50% of pregnancies. The FDA label still warns that even a single capsule, regardless of strength, taken during pregnancy can cause severe birth defects. There is no established safe threshold.
The exact day of exposure determined which structures were affected. A tablet taken on day 20 post-fertilization caused ear and brain malformations. Day 24: absent arms. Day 28: leg defects. Day 32: triphalangeal thumbs. The developing embryo was building its body on a precise schedule, and thalidomide was disrupting whichever structure was forming on the day the pill was swallowed. One pill, one day, one specific malformation.
The United States was largely spared. An FDA medical reviewer named Frances Oldham Kelsey rejected the application four times, insisting the safety data was insufficient. The manufacturer eventually gave up. Kelsey's stubbornness led directly to the 1962 Kefauver-Harris Amendment, which fundamentally strengthened FDA drug approval requirements. Modern drug regulation exists in part because of what thalidomide did.
The second life
This should have been the end of thalidomide. It wasn't.
In 1964, an Israeli dermatologist named Jacob Sheskin at Hadassah University Hospital in Jerusalem gave thalidomide to a 44-year-old man with severe leprosy complications - not for any scientific reason, but because it was the only sedative available in the hospital. After four tablets the patient's painful skin lesions dramatically improved within days. Withdrawal brought recurrence; reinstitution resolved it again. Sheskin treated six more patients with the same result and published the finding. Over the following decades, thalidomide was studied as an immunomodulatory drug.
In July 1998 - 37 years after it was pulled from markets worldwide - the FDA approved thalidomide (brand name Thalomid) for erythema nodosum leprosum (ENL), a painful inflammatory complication of leprosy. The approval came with the strictest safety controls ever placed on a drug - the THALOMID REMS program, designed to make fetal exposure impossible.
The leprosy finding hinted at something broader - thalidomide had immunomodulatory properties that nobody fully understood. That ambiguity attracted attention from cancer researchers. In the early 1990s, Harvard surgeon Judah Folkman was pioneering the theory that tumors need blood vessel growth (angiogenesis) to survive - and hypothesized the same was true of blood cancers. In 1994, a member of his lab, Robert D'Amato, discovered that thalidomide inhibited angiogenesis. Around that time, the wife of a man dying of myeloma called Folkman asking about his anti-angiogenesis work. Folkman persuaded the patient's oncologist, Bart Barlogie at the University of Arkansas, to try thalidomide. That led to a clinical trial whose results were published in the New England Journal of Medicine in 1999 - about a third of patients with relapsed multiple myeloma responded. In May 2006, the FDA granted accelerated approval for thalidomide in combination with dexamethasone for newly diagnosed multiple myeloma.
The Celgene franchise
By the 1990s, thalidomide's original patents had been expired for two decades. The molecule was in the public domain. But no pharmaceutical company would touch it - the name was toxic, the regulatory burden of preventing fetal exposure was enormous, and the science was uncertain. It took an unlikely company to see past the stigma.
Celgene wasn't even a pharmaceutical company when it started. Spun out of Celanese Corporation in 1986, it was originally a bioremediation company - engineering microbes to clean up toxic waste. In 1991, co-founder Sol Barer visited Rockefeller University to discuss a tuberculosis project. Immunologist Gilla Kaplan redirected the conversation: she had new data showing thalidomide could inhibit TNF-alpha, a key inflammatory cytokine. Celgene licensed the research rights in 1992, and by 1994 had abandoned bioremediation entirely to bet the company on the most reviled molecule in pharmaceutical history.
It was a bet nobody else was willing to make. Celgene built the rigorous safety infrastructure - the S.T.E.P.S. program (later formalized as the FDA-mandated REMS) - that made it possible to prescribe thalidomide safely. Pregnancy testing, prescriber certification, pharmacy controls, secure distribution. The system that made thalidomide usable again was as much an innovation as the drug itself.
Then Celgene's chemists did something more important: they took the thalidomide scaffold and engineered a better version. Lenalidomide (Revlimid) - more potent, less neurotoxic, and effective in a wider range of blood cancers - was a genuinely new molecule with its own composition-of-matter patents. Revlimid was approved in 2005 for myelodysplastic syndrome (5q-deletion MDS) and in 2006 for multiple myeloma. A third derivative, pomalidomide (Pomalyst), followed in 2013 for relapsed myeloma.
These drugs were called immunomodulatory drugs (IMiDs). The name was descriptive but vague - because nobody actually knew their precise molecular mechanism. They worked. They worked extremely well. But asking how they worked yielded hand-waving about cytokines and angiogenesis.
Revlimid became the backbone of myeloma treatment worldwide. Peak revenue hit $12.8 billion in 2021 - making it the second-highest-revenue cancer drug on the planet, behind only Keytruda. In 2019, Bristol-Myers Squibb acquired Celgene for $74 billion - one of the largest pharma deals ever - driven primarily by a franchise built on a molecule no other company would touch.
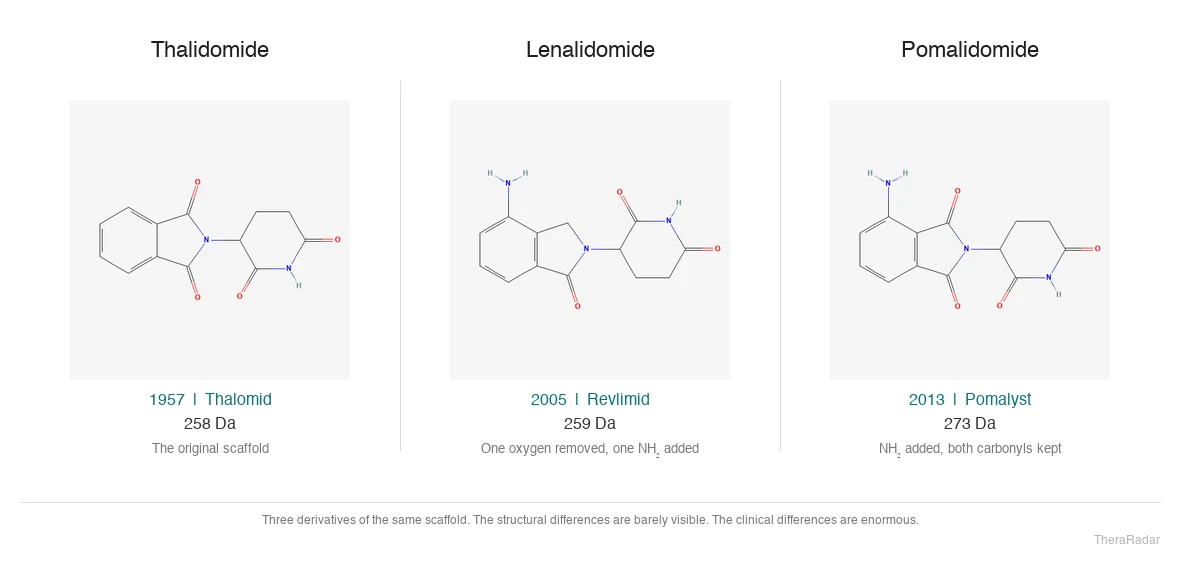
Structures from PubChem: thalidomide (CID 5426), lenalidomide (CID 216326), pomalidomide (CID 134780). Three derivatives of the same scaffold. Celgene turned two atoms of difference into a $12.8 billion franchise.
| Drug | Brand | FDA approval | Primary indication | Peak revenue |
|---|---|---|---|---|
| Thalidomide | Thalomid | 1998 (ENL), 2006 (MM) | Multiple myeloma | ~$300M |
| Lenalidomide | Revlimid | 2005 (MDS), 2006 (MM) | Multiple myeloma, MDS | $12.8B (2021) |
| Pomalidomide | Pomalyst | 2013 | Relapsed/refractory MM | ~$3.5B |
All three drugs are derivatives of the same thalidomide scaffold. Built by Celgene; acquired by BMS in 2019 for $74B.
Fifty years, no answer
From 1961 to 2010, scientists proposed dozens of theories for how thalidomide caused birth defects. Anti-angiogenesis. Oxidative stress. DNA intercalation. Nerve damage. None fully explained the pattern of limb malformations - and none explained why the drug was so effective in cancer.
The anti-angiogenesis theory was the most persistent. After D'Amato showed thalidomide inhibited blood vessel growth in 1994, it seemed to explain both the birth defects (developing limbs need blood vessels) and the cancer activity (tumors need blood vessels too). But it didn't explain the day-specific malformation pattern, and it didn't explain why lenalidomide - which has weaker anti-angiogenic activity - was far more potent against myeloma.
The problem was fundamental: nobody knew what protein thalidomide was actually binding to. Without a target, the mechanism was a black box. Three generations of chemists made better thalidomide analogs without understanding what they were optimizing.
Cereblon
In 2010, a team led by Hiroshi Handa at the Tokyo Institute of Technology published a paper in Science that solved the fifty-year mystery.
The approach was almost primitive. They coated tiny ferrite beads with thalidomide, dipped them into cell extracts, and pulled out whatever stuck. Mass spectrometry identified the protein: cereblon (CRBN).
CRBN turned out to be part of an E3 ubiquitin ligase complex - specifically, the CRL4CRBN complex (Cul4A-DDB1-CRBN-RBX1). E3 ligases are the cell's protein disposal system. They tag proteins with ubiquitin chains, marking them for destruction by the proteasome - the cell's molecular shredder.
The Handa team showed that thalidomide binding to CRBN disrupted its normal activity in zebrafish and chick embryos, leading to limb malformations and loss of Fgf8 expression. Fifty years of mystery, one paper in Science.
But the full picture was still incomplete. If thalidomide just blocked CRBN, that would make it an inhibitor - a familiar concept. What happened next was stranger.
The mechanism nobody expected
In late 2013 and early 2014, two groups published landmark papers in Science that completed the picture. Kronke et al. and Lu et al. showed that lenalidomide and its relatives don't just block CRBN. They change its surface.
When an IMiD binds to CRBN via its glutarimide ring, it alters the ligase's binding surface, creating a new protein-protein interface. This new surface grabs proteins that CRBN would never normally touch - neosubstrates. The key neosubstrates are the zinc finger transcription factors Ikaros (IKZF1) and Aiolos (IKZF3), which are essential for myeloma cell survival.
Once recruited, these neosubstrates are tagged with ubiquitin and destroyed by the proteasome. The drug doesn't block the target directly. It hijacks the cell's own waste disposal system to destroy proteins that were never supposed to be on the menu.
Scientists realized that thalidomide and its derivatives had been doing something genuinely new all along. They weren't inhibitors. They were molecular glues - small molecules that stabilize a protein-protein interaction between an E3 ligase and a target that the ligase would otherwise ignore. Revlimid was already a $12.8 billion drug before scientists worked out what it was actually doing inside a cell.
Go deeper with Pro
Pro gives you the data — patent cliffs, trial analytics, competitive landscapes, revenue tracking. Briefs tell the story with the data. Launch pricing: $99/month.
See Pro plansFrom accident to design
Thalidomide was a molecular glue by accident. But what if you could do this on purpose?
Two years before the cereblon discovery, in 2001, Yale chemist Craig Crews and Caltech biologist Raymond Deshaies had already proposed a different approach to the same idea. Their concept: build a bifunctional molecule with one end that binds a disease-causing protein and another end that recruits an E3 ligase. Connect the two with a chemical linker. The ligase tags the target protein with ubiquitin, the proteasome destroys it, and the bifunctional molecule is released to do it again.
They called these molecules PROTACs - Proteolysis-Targeting Chimeras. The first PROTAC (Protac-1) was crude - a peptide-based tool compound that targeted methionine aminopeptidase-2. It wasn't a drug. But the concept was radical: instead of blocking a protein's function, you could eliminate the protein entirely.
For the next decade, PROTACs were an academic curiosity. Too large, too peptide-like, not drug-like enough. Then the cereblon and neosubstrate discoveries changed the field. Once scientists understood that CRBN could be co-opted to degrade specific proteins - because thalidomide was already doing it - the E3 ligase handle became available for rational design.
Crews founded Arvinas in 2013 to turn PROTACs into drugs. Venture capitalists were skeptical - the molecules were 800-1,200 daltons, well beyond the 500-dalton ceiling that medicinal chemists treat as gospel. Crews argued the ceiling was a guideline, not a law of physics.
Two flavors of degradation
Targeted protein degradation now has two main branches, both rooted in the same insight.
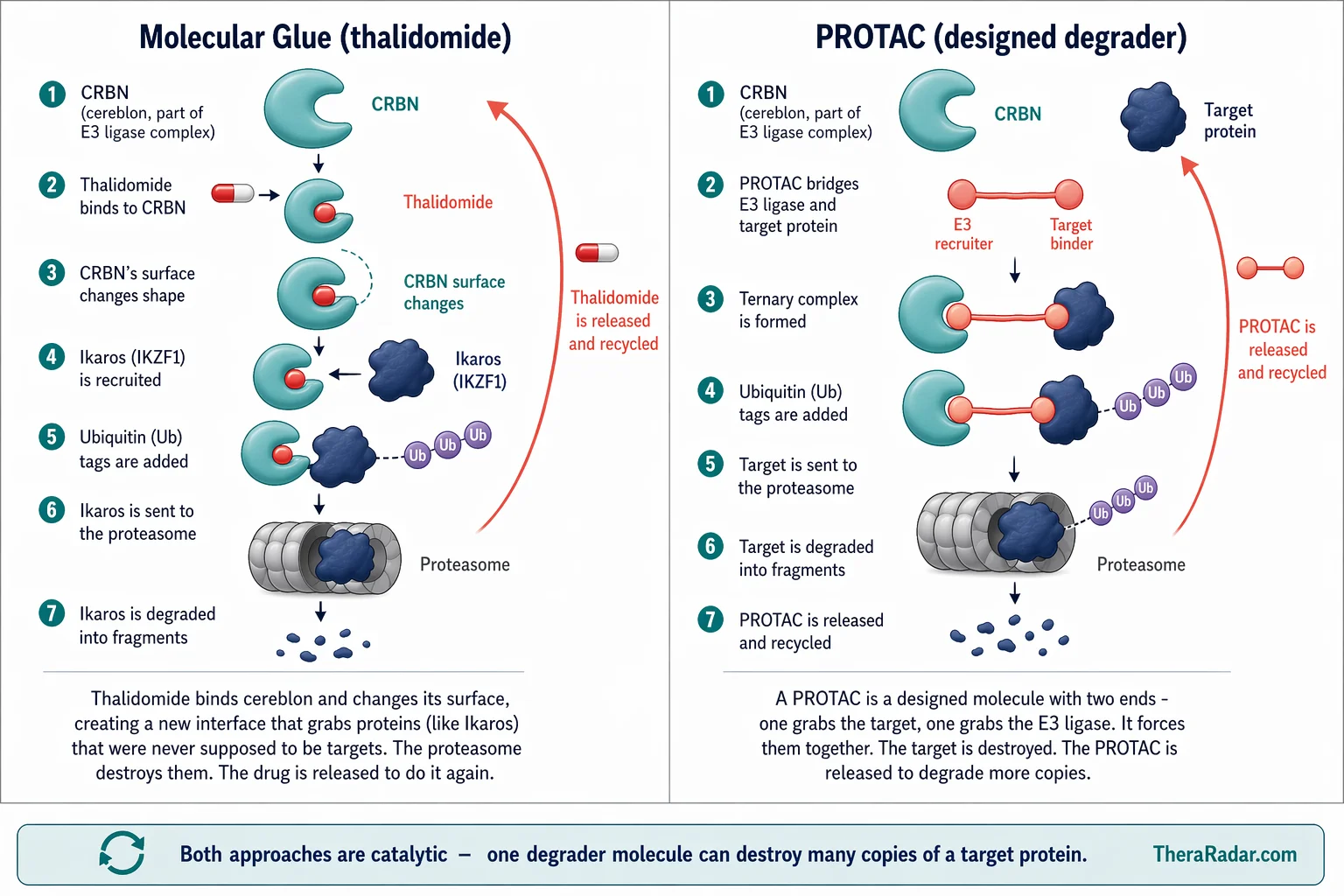
TheraRadar. Both approaches hijack the cell's protein disposal system. The molecular glue was discovered by accident. The PROTAC was designed on purpose.
| Molecular glues | PROTACs | |
|---|---|---|
| How it works | Binds E3 ligase, changes its surface to recruit neosubstrates | Bifunctional molecule physically bridges target and E3 ligase |
| Size | Small (<500 Da) - drug-like | Large (700-1,000+ Da) - beyond Lipinski's Rule of Five |
| Design approach | Historically serendipitous; rational design emerging | Rational: pick a target ligand + an E3 ligase recruiter + a linker |
| Oral bioavailability | Generally better (smaller) | More challenging (larger) |
| Catalytic? | Yes - recycled after each degradation event | Yes - released from ternary complex to degrade more targets |
| Key examples | Thalidomide, lenalidomide, iberdomide, mezigdomide | Vepdegestrant (ARV-471), BMS-986365 |
| Origin | Discovered accidentally (thalidomide, 1957) | Designed deliberately (Crews/Deshaies, 2001) |
Both approaches hijack the ubiquitin-proteasome system. The difference is how they bring the target and the E3 ligase together.
Molecular glues are small, drug-like molecules that bind an E3 ligase and change its surface to create a new protein-protein interaction with a neosubstrate. Thalidomide, lenalidomide, and pomalidomide are all molecular glues - they just weren't recognized as such until 2014.
PROTACs are larger, bifunctional molecules. One end binds the target protein. The other end recruits an E3 ligase (most commonly CRBN or VHL). A chemical linker connects the two. The PROTAC physically bridges the target and the ligase, forming a ternary complex. The target gets ubiquitinated and destroyed.
Both are catalytic - the degrader molecule is released after each event and can go destroy more copies of the target. This is fundamentally different from a traditional inhibitor, which must occupy the target protein one-to-one. A single PROTAC molecule can degrade many copies of a target protein over its lifetime.
Why it matters: the undruggable proteome
Traditional small-molecule drugs work by fitting into a protein's active site and blocking its function. The problem: only about 20% of human proteins have a well-defined binding pocket suitable for a conventional inhibitor. The other 80% - including many of the most important disease-driving proteins - are considered "undruggable" by conventional approaches.
Degraders sidestep this entirely. You don't need to block a protein's function - you need to eliminate the protein entirely. And for a PROTAC, you don't even need a high-affinity binding pocket on the target. You just need any handle - any small molecule that touches the target surface, even weakly - because the degradation mechanism is catalytic and sub-stoichiometric. Weak binders that would fail as conventional inhibitors can succeed as degrader warheads.
Transcription factors like Ikaros - proteins without defined active sites that couldn't be touched by conventional drugs - were the first proof. Thalidomide was already degrading them. The field's job was to do it on purpose, for targets of its choosing.
The landscape in 2026
Targeted protein degradation has gone from an academic concept to one of the most active areas in drug development. Dozens of degrader candidates are now in clinical trials across over a hundred active studies, spanning oncology, immunology, and neuroscience. BMS alone is running 69 degrader trials across its three CELMoD molecules.
| Drug | Type | Target | Company | Indication | Trials | Most advanced |
|---|---|---|---|---|---|---|
| Vepdegestrant | PROTAC | Estrogen receptor | Arvinas / Pfizer | ER+/HER2- breast cancer | 24 | FDA approved (May 2026) |
| Iberdomide | CELMoD | CRBN neosubstrates | BMS (ex-Celgene) | Multiple myeloma, lymphoma | 35 | Phase 3 |
| Mezigdomide | CELMoD | CRBN neosubstrates | BMS (ex-Celgene) | Relapsed/refractory MM | 19 | Phase 3 (positive data) |
| Golcadomide | CELMoD | CRBN neosubstrates | BMS (ex-Celgene) | Lymphoma | 15 | Phase 3 |
| BGB-16673 | PROTAC | BTK | BeiGene | B-cell malignancies | 10 | Phase 3 |
| NX-5948 | PROTAC | BTK | Nurix | CLL/SLL, B-cell lymphoma | 7 | Phase 2 (pivotal) |
| BMS-986365 | PROTAC | Androgen receptor | BMS | Prostate cancer | 4 | Phase 3 |
| KT-621 | PROTAC | STAT6 | Kymera | Atopic dermatitis | 4 | Phase 2 |
| ARV-766 | PROTAC | Androgen receptor | Arvinas / Novartis | Prostate cancer | 1 | Phase 1/2 |
| ARV-806 | PROTAC | KRAS G12D | Arvinas | KRAS G12D solid tumors | 1 | Phase 1/2 |
Trial counts from ClinicalTrials.gov via TheraRadar (May 2026). Excludes legacy IMiDs (lenalidomide, pomalidomide). CELMoD = cereblon E3 ligase modulator.
Vepdegestrant (brand name Veppanu), developed by Arvinas and partnered with Pfizer, crossed the finish line first. It degrades the estrogen receptor in breast cancer - particularly in tumors with ESR1 mutations that make them resistant to conventional anti-estrogen therapies. The FDA approved it in May 2026, making it the first purpose-built PROTAC to reach patients.
Meanwhile, BMS is building the next generation of molecular glues - what they call CELMoDs (cereblon E3 ligase modulators). Iberdomide, mezigdomide, and golcadomide are all in Phase 3, each targeting different CRBN neosubstrates for blood cancers. In March 2026, mezigdomide showed positive Phase 3 results in relapsed myeloma - the first positive Phase 3 for a next-generation CELMoD. Between these three molecules, BMS has 69 active degrader trials - more than any other company. The direct line from Celgene's thalidomide bet to BMS's CELMoD portfolio is unmistakable.
Two BTK degraders - BeiGene's BGB-16673 (Phase 3) and Nurix's NX-5948 (pivotal Phase 2) - are advancing in B-cell cancers, competing to degrade a target that conventional BTK inhibitors can only block. And Arvinas, the company Crews founded, now has three PROTACs in the clinic: vepdegestrant (estrogen receptor), ARV-766 (androgen receptor, partnered with Novartis), and ARV-806 (KRAS G12D - the same "undruggable" target that took 40 years to crack with conventional inhibitors).
- 1957 Thalidomide launched in Germany as an over-the-counter sedative
- 1961 Withdrawn after 10,000+ birth defects. Most infamous drug in history.
- 1998 FDA approves thalidomide for leprosy (ENL) - 37 years after withdrawal
- 2001 Crews and Deshaies publish first PROTAC concept (PNAS)
- 2005 Celgene's lenalidomide (Revlimid) approved - engineered thalidomide derivative
- 2010 Handa lab identifies cereblon as thalidomide's target (Science) - 50-year mystery solved
- 2014 Kronke et al. discover neosubstrate degradation mechanism - thalidomide was a molecular glue
- 2019 BMS acquires Celgene for $74B, largely driven by Revlimid
- 2021 Revlimid hits $12.8B peak revenue - #2 cancer drug worldwide
- 2026 Vepdegestrant approved. First purpose-built PROTAC reaches patients - 65 years after thalidomide was pulled.
The longest arc in drug development
About 2,400 thalidomide survivors are still alive today. The drug that harmed them also forced the creation of the 1962 Kefauver-Harris Amendment - the reason every drug now requires proof of safety and efficacy before reaching patients. Modern drug regulation is, in part, a direct consequence of what thalidomide did to those 10,000 children.
And the molecule itself turned out to be doing something nobody was looking for: hijacking the cell's own protein disposal system to selectively destroy specific proteins. A mechanism misunderstood for fifty years. A franchise that generated $12.8 billion in peak annual revenue before scientists worked out how it functioned. And now, a design principle - targeted protein degradation - with over a hundred active clinical trials aimed at the 80% of human proteins that conventional drugs can't touch.
Vepdegestrant's approval in May 2026 closed the loop. Sixty-five years from the worst accident in drug history to a rationally designed therapy built on the same molecular principle. The longest arc in pharmaceutical development, from a sleeping pill that should never have been sold to a cancer drug that couldn't have been built without understanding what went wrong.
Get the next Brief in your inbox — free
Every drug has multiple stories. Most never get told. One brief every week. No spam.
Archive at /briefs/. Unsubscribe anytime.
Sources
- Thalidomide teratogenesis and critical window: Vargesson - "Thalidomide-induced teratogenesis: history and mechanisms," Birth Defects Research (2015)
- FDA label (single-dose warning): DailyMed - THALOMID prescribing information
- Sheskin's leprosy case (1965): Sheskin J - "Thalidomide in the treatment of lepra reactions," Clinical Pharmacology & Therapeutics 6(3):303-306
- Thalidomide in myeloma (1999): Singhal et al. - "Antitumor activity of thalidomide in refractory multiple myeloma," NEJM 341(21):1565-1571
- Frances Oldham Kelsey: National Women's History Museum - Frances Kathleen Oldham Kelsey
- Cereblon discovery (2010): Ito, Ando, Handa et al. - "Identification of a primary target of thalidomide teratogenicity," Science 327(5971):1345-1350
- Neosubstrate mechanism (2014): Kronke et al. - "Lenalidomide causes selective degradation of IKZF1 and IKZF3," Science (2014)
- First PROTAC concept (2001): Sakamoto, Crews, Deshaies - "Protacs: chimeric molecules that target proteins for ubiquitination and degradation," PNAS 98(15):8554-8559
- Molecular glue mechanisms: PMC - Molecular mechanisms of action of thalidomide and its derivatives (2020)
- Molecular glues review: PMC - Molecular Glues: The Adhesive Connecting TPD to the Clinic (2023)
- Vepdegestrant NDA: Arvinas - FDA acceptance of NDA for vepdegestrant
- Mezigdomide Phase 3: BMS - Positive Phase 3 results from SUCCESSOR-2 (March 2026)
- PROTAC clinical landscape: Biopharma PEG - PROTAC Clinical Trials 2025 Update
- Thalidomide StatPearls: NCBI StatPearls - Thalidomide
Spot an error? Reach out at hello@theraradar.com.